
年度综述:T细胞与NK细胞在肿瘤免疫中的协同作用及治疗策略
摘要:T细胞和自然杀伤(NK)细胞在肿瘤免疫中互补,其双重攻击有望深化免疫疗法成效。NK 细胞能募集树突状细胞(DC)入肿瘤,增强CD8 T细胞反应,且T细胞分泌的IL-2可激活NK细胞。肿瘤细胞存在免疫逃避机制,如针对NKG2D受体及其配体的逃避,而T细胞和NK细胞共享抑制性与激活性受体,如CD161-CLEC2D、TIGIT-CD155和NKG2A/CD94-HLA-E等,靶向这些受体可增强免疫力。此外,基于抑制性和激活性细胞因子的治疗策略对肿瘤内两种淋巴细胞群的功能影响深远。
一、肿瘤异质性是肿瘤免疫疗法面临的挑战
肿瘤细胞异质性是肿瘤治疗的难题,一方面新的突变催生耐药肿瘤亚克隆的产生;另一方面肿瘤细胞能改变细胞状态来适应治疗,如在遇到杀伤药物后肿瘤细胞可转入静止态。肿瘤特异性CD8 T细胞靠TCR识别MHC-I呈递的肿瘤多肽来杀伤肿瘤细胞,活化的T细胞通过分泌IFN-γ上调 MHC-I抗原呈递相关基因,使肿瘤细胞对T细胞攻击敏感,但T细胞识别依赖肿瘤细胞MHC-I持续表达,一旦肿瘤细胞因突变或表观遗传机制失去MHC-I的表达,T细胞就无法识别。鉴于T细胞与NK细胞识别肿瘤细胞策略不同,二者可以协同攻击肿瘤细胞,NK细胞可杀逃避T细胞识别的肿瘤细胞,MHC-I缺失会使肿瘤细胞对NK细胞更敏感,因为几种MHC-I蛋白是NK细胞上抑制受体的配体。
二、T细胞和NK细胞对肿瘤免疫的互补作用
在人类与实验动物模型中,T细胞介导免疫的重要性显著,FDA批准的诸多疗法旨在增强其介导的免疫力,像阻断T细胞抑制性受体的PD-1和CTLA-4抗体,以及基因工程T细胞疗法。细胞毒性T细胞固然在肿瘤免疫中发挥着关键作用,不过CD4 T细胞同样关键,在非免疫原性肉瘤模型中,仅有CD8 T细胞的MHC-I新抗原还不够,需同时有MHC-II新抗原才能使肉瘤对联合检查点阻断有反应,CD4 T细胞既能初始化 CD8 T细胞毒性状态,还能重编程肿瘤微环境中的髓系细胞,使其处于更具免疫刺激性的状态。
NK 细胞通过种系编码激活受体识别肿瘤细胞,大多数循环的人类NK细胞处于稳定的细胞毒性效应状态(CD16+CD56dim细胞),所以NK细胞能在抗肿瘤免疫早期响应。而T细胞的反应则会延迟,因为T细胞需从少量前体细胞大量扩增克隆。NK细胞在防止肿瘤细胞转移扩散上也作用重大,在小鼠体内实验中,NK细胞的耗竭会大大增加自发转移的数量。在血液恶性肿瘤治疗中,NK细胞的抗体依赖性细胞毒性是抗体治疗的重要效应机制,如利妥昔单抗。ADCC主要由 NK细胞通过活化的CD16A受体(由 FCGR3A 编码)介导,该受体可识别肿瘤细胞结合的IgG抗体,临床试验的基因学证据直接表明,CD16A受体与此类抗肿瘤单抗的治疗活性有关。
三、T 细胞介导的肿瘤免疫中的NK细胞-树突状细胞轴
NK细胞不仅有细胞毒性,还能招募DC进入实体瘤助力T细胞介导的肿瘤免疫(图1)。传统DC分cDC1和cDC2亚群,cDC1善于摄取凋亡肿瘤细胞并转运相关抗原至肿瘤引流淋巴结,还能招募激活肿瘤特异性CD8 T细胞。在Batf3基因敲除小鼠中,缺失cDC1会使保护性抗肿瘤免疫失效。NK细胞在招募cDC1进入肿瘤方面发挥着关键作用,研究发现NK细胞分泌趋化因子如XCL1、XCL2和CCL5招募cDC1,且前列腺素E2会抑制该招募;NK细胞内会表达FLT3, FLT3配体与cDC1浸润及患者生存期相关,NK 细胞耗竭会减少cDC1招募。在人类黑色素瘤等癌症中,cDC1和NK细胞在肿瘤中的浸润密切相关。这些研究证明了肿瘤中NK细胞和T细胞反应之间的重要联系:NK细胞招募并支持DCs的存活,而DCs又是T细胞介导的肿瘤免疫的关键。
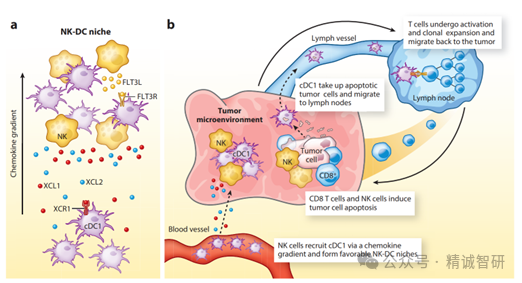
图1. NK细胞募集树突状细胞进入肿瘤
(a) NK细胞与肿瘤中的cDC1形成的细胞微环境。交叉呈递的cDC1表达XCR1趋化因子受体,从而被分泌XCL1和XCL2的NK细胞招募。NK细胞还会分泌FLT3L,诱导树突状细胞分化。(b) NK细胞招募的cDC1对CD8和CD4 T细胞与细胞相关抗原的启动非常重要。cDC1从死亡的肿瘤细胞中摄取凋亡片段,并将这些抗原运送到肿瘤引流淋巴结,在那里激活CD8和CD4 T 细胞。因此,NK细胞有助于诱导T细胞介导的抗肿瘤免疫。
四、T细胞与NK细胞识别肿瘤细胞的分子逻辑
T细胞通过TCR识别MHC结合肽抗原来识别肿瘤细胞,TCR掌控T细胞关键功能,肿瘤下调或丧失MHC-I表达就可逃避CD8 T细胞免疫(图2a)。而NK细胞用一系列种系编码的激活和抑制受体识别受压和转化的细胞,没有一种受体是识别肿瘤细胞所必需的,其细胞毒性由多受体信号整合控制(图2b),比T细胞识别更灵活,因此,二者对肿瘤细胞施加不同选择压力,为双重靶向肿瘤提供依据。
TCR激活受到一系列的共刺激和共抑制受体的调节。CD28共刺激受体在肿瘤特异性T细胞启动起核心作用,它能与活化DC表面表达的CD80和CD86配体结合。共抑制受体CTLA-4和 PD-1则被设计成主要的药物靶点,在转移性黑色素瘤治疗中有显著生存获益,且PD-1或其配体PD-L1靶向抗体已用于多种癌症。这些受体表达的时间和空间模式决定其独特作用,如CTLA-4在TCR 触发后转运至细胞表面,并减弱APC对T细胞的早期激活。相反,PD-1在肿瘤等部位经 TCR 重复激活后高表达,重要的是,活化的T细胞释放的IFN-γ会高度上调肿瘤细胞中PD-L1的表达,从而形成一种称为适应性抵抗的负反馈机制。LAG-3是T细胞第三个抑制性受体,在小鼠肿瘤模型中,LAG-3抗体可增强 PD-1阻断的疗效,与PD-1单药治疗相比,LAG-3和PD-1单抗的联合治疗可带来显著的生存获益。
NK细胞通过NKG2D、NKp46、NKp30 和 NKp44受体识别肿瘤细胞(图2b),NKG2D与DNA损伤和cGAS-STING信号在肿瘤细胞中诱导的配体结合;NKp46识别内质网应激误定位的钙网蛋白;人类NKp30识别肿瘤细胞系表达的B7-H6蛋白;NKp44受体可识别PDGF-DD,这是一种由肿瘤细胞释放的促进血管生成的生长因子。PDGF-DD与NKp44接合会引发IFN-γ和TNF-α 的分泌,从而抑制肿瘤细胞的生长。同时,MHC-I蛋白是NK 细胞抑制性受体配体,MHC-I 缺失使肿瘤细胞对NK细胞杀伤敏感,为同时使用T细胞和NK细胞的免疫疗法策略提供了重要依据(图2b、c)。
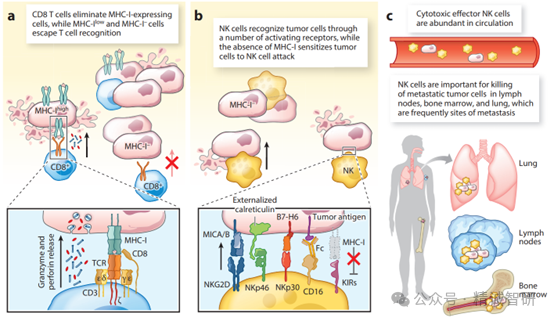
图2. CD8 T细胞和NK细胞在肿瘤免疫中的互补作用
(a) MHC-I蛋白向CD8 T细胞展示肿瘤肽,导致T细胞活化并释放穿孔素和颗粒酶,诱导肿瘤细胞凋亡。CD8 T细胞会施加强大的选择压力,导致不再被CD8 T细胞识别的MHC-I缺失细胞的生长。(b) NK细胞通过一组种系编码的激活受体(包括NKG2D、NKp46、NKp30和CD16)识别肿瘤细胞。NK细胞对肿瘤细胞的活性会因MHC-I蛋白的抑制性受体(包括人类的抑制性KIRs)而减弱。因此,缺乏MHC-I的肿瘤细胞更容易被NK细胞杀死。(c) NK细胞对转移性肿瘤细胞的作用。NK细胞大量存在于血液、骨髓和肺部血管周围空间,肺部是肿瘤转移的常见部位。大多数循环中的NK细胞处于细胞毒性效应状态,可快速杀死迁移的肿瘤细胞。缩写:KIR:杀伤细胞免疫球蛋白样受体;TCR:T细胞受体。
五、代表治疗机会的T细胞和NK细胞共有受体
T细胞和NK细胞生物学经常被呈现为适应性和先天性识别的二分法。然而,NK细胞和CD8 T细胞的受体-配体系统中也存在相当大的重叠。因此,由CD8 T细胞和NK细胞共表达的激活和抑制性受体在抗肿瘤免疫中同时参与两个细胞毒淋巴细胞群体,增强了机体的抗肿瘤免疫应答。主要细胞受体-配体系统有抑制性CD161受体及其配体CLEC2D、抑制性NKG2A/CD94受体及其配体HLA-E、CD226受体及其抑制性对应物TIGIT、CD96和PVRIG、NKG2D受体及其应激诱导的配体等。
抑制性CD161受体及其配体CLEC2D:CD161受体属C型凝集素受体家族,它最初被鉴定为是NK细胞抑制受体,后发现也是肿瘤浸润T细胞的重要抑制受体。CLEC2D在多种人类肿瘤细胞与浸润髓样细胞表达,起源于生殖中心B细胞的B细胞淋巴瘤可表达高水平的 CLEC2D。单细胞RNA-seq显示多种肿瘤浸润T细胞高表达KLRB1(编码 CD161),胶质母细胞瘤中克隆扩增 CD8 T细胞KLRB1表达更高,且与细胞毒性基因表达相关。实验表明靶向该抑制性受体可增强T细胞和NK细胞抗肿瘤活性,如KLRB1编辑或用CD161阻断单抗处理T细胞,能增强杀伤力与细胞因子分泌,在人源化胶质母细胞瘤小鼠模型中有生存优势(图3a)。
抑制性NKG2A/CD94受体及其配体HLA-E:HLA-E是非经典MHC-Ib分子,与NK细胞和CD8 T细胞亚群的抑制性 NKG2A/CD94 受体结合,是NK细胞 “缺失自我” 识别模式的一部分,肿瘤细胞常过度表达HLA-E,多数循环和肿瘤浸润 NK细胞持续表达NKG2A/CD94受体,部分肿瘤浸润CD8 T细胞也上调其表达。在多种小鼠肿瘤模型研究中发现,活化T细胞分泌的IFN -γ会上调肿瘤细胞表面Qa-1(鼠类HLA-E同源物),NKG2A阻断单抗可增强疫苗疗效,人源化NKG2A阻断抗体莫那利珠单抗正在临床开发,与西妥昔单抗等联合治疗有一定效果,还需三期临床试验深入研究(图3c)。
CD226受体和抑制性对应物TIGIT、CD96和PVRIG:CD226是NK细胞和 CD8 T细胞的激动受体,CD226缺陷小鼠肿瘤生长加速。其配体CD155在人类肿瘤细胞广泛表达,CD226还能结合CD112。TIGIT、CD96和PVRIG是拮抗 CD226的抑制受体,TIGIT研究最多,在多种肿瘤内浸润CD8 T细胞表面高表达,也是NK细胞重要抑制受体,抑制TIGIT和PD-L1可增强肿瘤免疫力。TIGIT阻断性抗体替瑞利尤单抗的2期临床试验显示联合疗法提高NSCLC 患者总体反应率,目前在3期临床试验评估。对CD96和PVRIG研究较少,小鼠模型中使它们失活可增强抗肿瘤免疫力,靶向PVRIG的单抗正在 1 期临床试验评估(图3b)。
NKG2D受体及其应激诱导配体:NKG2D受体由NK细胞、CD8 T细胞等表达,连接后诱导穿孔素依赖性细胞溶解,为CD8 T细胞提供共刺激信号,能识别受压、转化细胞上调的配体,如人类的MICA/MICB和ULBP1- 6蛋白等。在多种肿瘤中能检测到NKG2D配体表达,缺乏NKG2D的小鼠肿瘤易感性增强。肿瘤细胞会通过ERp5等使MICA/B蛋白质解体脱落逃避免疫攻击,用针对 MICA/B α3结构域的单抗可抑制脱落,诱导NK细胞杀伤肿瘤细胞,对MHC - I缺失的转移瘤也有活性(图4a)。
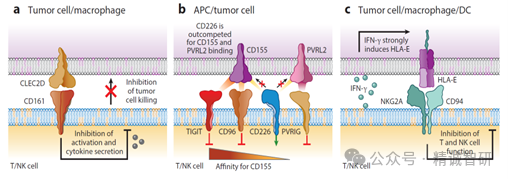
图3.T 细胞和 NK 细胞共有的抑制受体
(a) NK细胞表达CD161抑制受体,肿瘤浸润的CD8和CD4 T细胞高度上调其表达。它与CLEC2D配体结合,肿瘤细胞和肿瘤微环境中的免疫细胞均可表达该配体。CD161 信号可抑制NK细胞和CD8 T细胞的细胞毒活性,还可抑制这两种淋巴细胞群产生细胞因子。(b) CD226激活受体受NK细胞和T细胞中三种抑制受体的调节:TIGIT、CD96和PVRIG。CD226与CD155结合,CD155是一种在多种人类癌症类型中高度表达的蛋白质。与CD226相比,TIGIT和CD96与CD155结合的亲和力更高,因此在配体结合方面会超过激活受体。PVRIG抑制受体的主要配体是PVRL2。(c) 抑制性 NKG2A/CD94受体由NK细胞和一部分CD8 T细胞表达。它与HLA-E结合,而HLA-E 的表达是由活化的T细胞和NK细胞分泌的IFN-γ诱导的。缩写:APC,抗原递呈细胞。
六、一种诱导T细胞和NK细胞双重攻击耐药性肿瘤的癌症疫苗
多数肿瘤疫苗聚焦肽表位,需个性化,且细胞毒性T细胞致肿瘤克隆出现MHC-I缺失,使肿瘤细胞对T细胞不可见。引发T细胞和NK细胞双重攻击的疫苗可防MHC-I 缺失型肿瘤克隆,如针对参与MICA/B蛋白水解脱落的α3结构域的疫苗,可诱导高滴度抗体,抑制脱落、增加配体密度,使T细胞和NK细胞涌入肿瘤,在切除原发肿瘤后免疫接种能减少转移瘤生长,对MHC-I缺失肿瘤免疫需NK细胞和CD4 T细胞,CD4 T细胞招募NK细胞,NK细胞成为关键效应细胞,疫苗诱导的抗体还能增强DC 交叉呈递抗原与NK细胞杀伤力,确保此类疫苗中含有CD4 T细胞表位非常重要,因为它们能促进CD8 T细胞分化为细胞毒性效应细胞状态,对TME中的免疫抑制髓系细胞进行重编程,并诱导NK细胞的招募(图4b)。
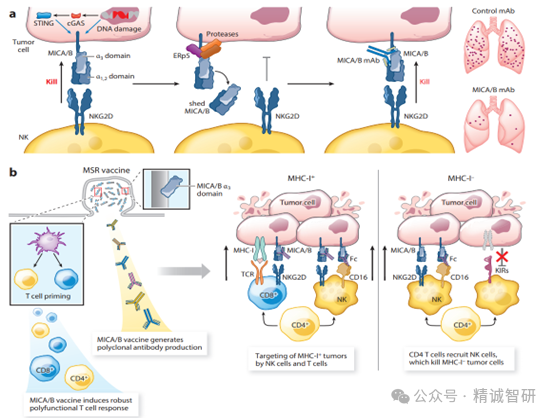
图4.NKG2D-MICA/B 通路的治疗靶点
(a)活化的 NKG2D 受体由人类NK细胞、CD8 T细胞、NKT 细胞和 γδ T细胞表达。它能与肿瘤细胞上的应激诱导配体 MICA 和 MICB(MICA/B)结合,但肿瘤会通过 ERp5 二硫异构酶和蛋白酶 ADAM10 及 ADAM17 的协同作用使 MICA/B 蛋白质解体脱落,从而逃避 NKG2D 介导的免疫攻击。针对 MICA/B α3 结构域的单克隆抗体可抑制蛋白水解脱落,并诱导 NK 细胞介导的针对转移灶的免疫攻击。(b) 以 MICA/B α3 结构域为靶点的疫苗可诱导针对这种应激蛋白的 T 细胞反应以及抑制 MICA/B 脱落的多克隆抗体。这种疫苗通过 CD4 T 细胞和 NK 细胞的协调作用,对 MHC-I缺失的肿瘤细胞保持活性。CD4 T细胞将 NK 细胞募集到肿瘤中,而NK细胞通过NKG2D和CD16受体激活后杀死肿瘤细胞。
七、靶向抑制性细胞因子和小分子介质以增强T细胞和NK细胞的功能
TGF-β:是免疫抑制细胞因子,抑制肿瘤内T细胞和NK细胞功能,抑制T细胞增殖与效应功能,在NK细胞中抑制关键代谢程序、激活受体表达。因TGF -β以非活性前体沉积在细胞外基质,靠整合素α(v)受体释放活性形式,上皮癌细胞高表达整合素αvβ6,抗体靶向其为抑制上皮癌中TGF -β激活的方法,一种整合素αvβ6/8单抗能增强T细胞杀伤力、使肿瘤对PD-1抑制剂敏感;还可阻断TGF-β1从潜伏复合物激活,联合PD-1单抗使小鼠肿瘤模型获生存获益且无心脏瓣膜损伤副作用。
前列腺素E2(PGE2):有免疫抑制与促癌作用,环氧化酶-1和2合成PGE2,在部分人类癌症常过度表达。抑制小鼠黑色素瘤细胞系PGE2合成,使肿瘤微环境细胞因子转变,CD103+ cDC1增加、NK细胞浸润增多,Ptgs1/2- /-肿瘤在免疫健全小鼠自发排斥,用阿司匹林与PD-1阻断联合抑制COX可减缓肿瘤生长,人类黑色素瘤中PTGS2 (COX-2)表达与细胞因子相关,长期服阿司匹林与结肠癌发病率降低有关,林奇综合征患者服阿司匹林可降低结肠癌风险。
八、改良刺激性细胞因子以促进T细胞和NK细胞介导的肿瘤免疫
有几种主要的细胞因子作用于T细胞和NK细胞,包括IL-2、IL-12、IL-15和IL-18,为增强T细胞和NK细胞功能提供了机会。
IL-2:有多种变体设计思路,其受体由IL-2Rβ和γ共链组成中度亲和性受体,表达于效应T细胞和NK细胞,IL-2Rα/CD25与它们结合成高亲和力受体,CD25在 Foxp3+ CD4 Tregs高表达,IL-2受限时Tregs优先扩增。设计的IL-2变体(super2)能激活效应细胞中度亲和性受体且不结合CD25;NKTR-214是聚乙二醇化的IL-2,能缓慢释放,增加肿瘤浸润T细胞和NK细胞数量,1期临床试验显示其可使患者病情稳定、淋巴细胞增殖,肿瘤活检基因表达与细胞积累改变。
IL-15:能激活效应T细胞和NK细胞但不激活Tregs,独特作用机制是经DC 等转呈,激活IL-2Rβγ复合物。开发的IL-15 超级拮抗剂ALT-803,融合相关结构域增强活性、延长半衰期,1期临床试验在部分血液恶性肿瘤复发患者产生治疗反应,诱导NK细胞和CD8 T细胞增殖、增强活化受体与颗粒酶B表达。
IL-18:是IL-1家族成员,被Caspase-1激活后通过MYD88和NF-κB 信号激活T细胞和NK细胞,IL-18Rα在NK细胞与肿瘤浸润T细胞上调,但受IL-18 结合蛋白负调控。开发的IL-18诱饵抗性变体DR-18,在多种小鼠肿瘤模型有治疗活性,使瘤内T细胞进入效应状态、减少衰竭标志物,诱导NK细胞活化,对 MHC- I缺乏肿瘤有效。
九、激活T细胞和NK细胞的联合疗法
在小鼠模型发现由抗肿瘤抗体(A)、半衰期延长的 IL-2(I)、抗PD-1(P)和多肽疫苗(V)组成的四药联合疗法 AIPV,可简化为单剂AIP后用PD-1加CTLA-4单抗进行免疫检查点阻断(ICB)。单剂量AIP治疗早期,NK细胞作用突出,巨噬细胞也有贡献,包括促炎趋化因子和细胞因子的表达上调。在AIP治疗前消耗NK细胞时,DC和效应T细胞招募所需的因子的上调表达大大降低。早期免疫激活还使cDC1摄取肿瘤抗原增加、活化状态提升,表明肿瘤结合抗体(A)与延长半衰期的IL-2(I)有助于早期NK细胞活化,进而招募DC和CD8 T细胞,说明利用T细胞和NK细胞共享途径可设计联合疗法。此外,先天性免疫刺激物如STING激动剂能诱导CD8 T细胞和NK细胞抗肿瘤反应,与IL-2超级激酶协同作用显著, 在挑战MHC-I缺失和MHC-I阳性的小鼠肿瘤中显示出强大的协同作用。
十、总结和未来方向
最近的发现表明,肿瘤中T细胞与NK细胞生物学特性紧密相关,NK细胞在T细胞介导的肿瘤免疫中作用关键,能招募DC并提支持其存活的生态位。二者虽分属先天与适应性免疫分支,但共享激活和抑制受体配体系统,为免疫疗法带来契机。未来研究将探究肿瘤中DC、NK细胞和T细胞形成的微环境优势,明确抑制其招募与功能的分子机制,新兴空间技术有望揭示它们的动态相互作用,助力发现免疫疗法新靶点,开发联合疗法。